bitBites (2026-02-11): Codex demo, AlphaGenome
R, data science and AI
- Codex: Two Codex tutorial posts caught my interest this week. Although I have no hands-on experience with popular coding agents (other than the shiny assistant from Positron), I would personally choose Codex over Claude Code purely for economic reasons. Stephen Turner demonstrates refactoring R package
{qqmam}1 with a single prompt2. The rednote post provides a step-by-step guide to setting up a “vibe coding” workflow in Codex Desktop.
dplyr column recording: extension of
case_whenwith cleaner coding.replace_when(x, .. ~ .., ...)lets you pull the primary input to the front (even allow pipe, although i dont like pipe inside function), making the intent more clear. and preserve the levels when it is a factor.record_values()kicks in whenever you use%in%or==in the condition side. It contains 2 APIsrecord_values(x, ..~.., ...)andrecord_values(x, from, to)with lookup list (created bytribble).replace_values(), same asreplace_when(x, .. ~ .., ...)when you just want to update original values. The syntax is same asrecord_values().
R packages of interest
{annotaR}: It annotates gene with 3 ontology system (GO term, associated disease, associated drug), supporting Open Target API for gene-disease and gene-drug annotation.
Baysien framework NIMBLE: {nimbleQuad} provides quadrature-based inference methods that use Laplace and Adaptive Gauss-Hermite Quadrature (AGHQ) approximation. {NIMBLE} v1.4.0 allows using “derived quantities” (eg. posterior mean, posterior variance) to calculate or record additional quantities of interest during an MCMC.
AlphaGenome
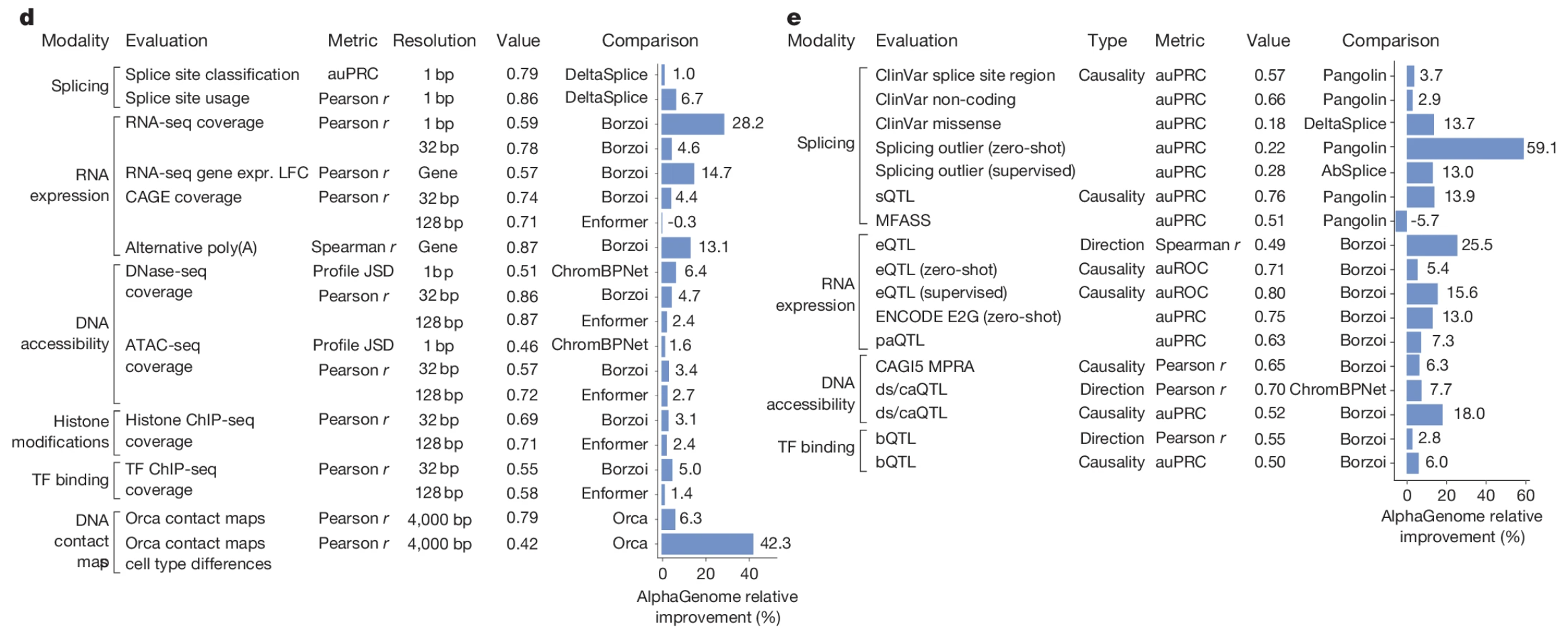
Google DeepMind published this foundation model AlphaGenome at Nature last week. Although it hss been on BioRxiv for over half a year, the release of peer-reviewed version still made a big slash in the field. This unified DNA model allows up to 1Mbp sequence input and predicts genome tracks for diverse modalities, including splicing (donor-acceptor sites, coverage, intron junction), gene expression, accessibility (ATAC, DNAase), ChIP-seq (histone, TF binding) and chromatin contact map, at as high as single nucleotide resolution. This single nucleotide resolution also enables the variant regulatory effect prediction, such as eQTL, caQTL/dsQTL, bQTL and paQTL, which is particularly impactful for rare variants where traditional association-based study lacks statistical power. Since it was trained on thousands of transcriptomic and epigenomic datasets across diverse cell types (cell lines and primary tissues), the model allows cell-specific prediction. Although paper admits the limitation of cell-specific prediction in certain tasks (eg. alternative splicing, expression fold change), this is a big breakthrough in my perspective. The cell context is critical to DNA regulatory landscape, which is frequently overlooked by previous tools. Through systematic benchmarking, the authors claims that integration of AlphaGenome scores from multiple modalities through shallow models (eg. random forest, LASSO) increase the performance of certain tasks. This is not surprising given the intrigated property of regulatory machinary represented by those genome tracks.
Below represents all the tasks that AlphaGenome has been benchmarked on. Github repo provides python API for non-commercial usage. Pre-trained model weights can be accessed through Hugging Face.
Footnotes
Turner, S. D. (2018). qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. Journal of Open Source Software, 3(25), 731. https://doi.org/10.21105/joss.00731.↩︎
OpenAI Codex App by Stephen Turner. https://blog.stephenturner.us/p/openai-codex-app-qqman↩︎