bitBites (2026-01-14): single-cell for drug development and clinical translation, R+Python polyglot, claude code
In this week’s bitBits, I will highlight recent advances in using single-cell transcriptomics for drug prioritization and clinical translation, including frameworks like DrugReflector, scFOCAL, and Pertpy that connect perturbation data with disease phenotypes and multicellular programs. On the tooling side, I share updates on polyglot workflows across R and Python and practical visualization tips, and reflect on agentic coding assistants like Claude Code.
Bioinformatics and biology
Drug prioritization by single cell transcriptomics
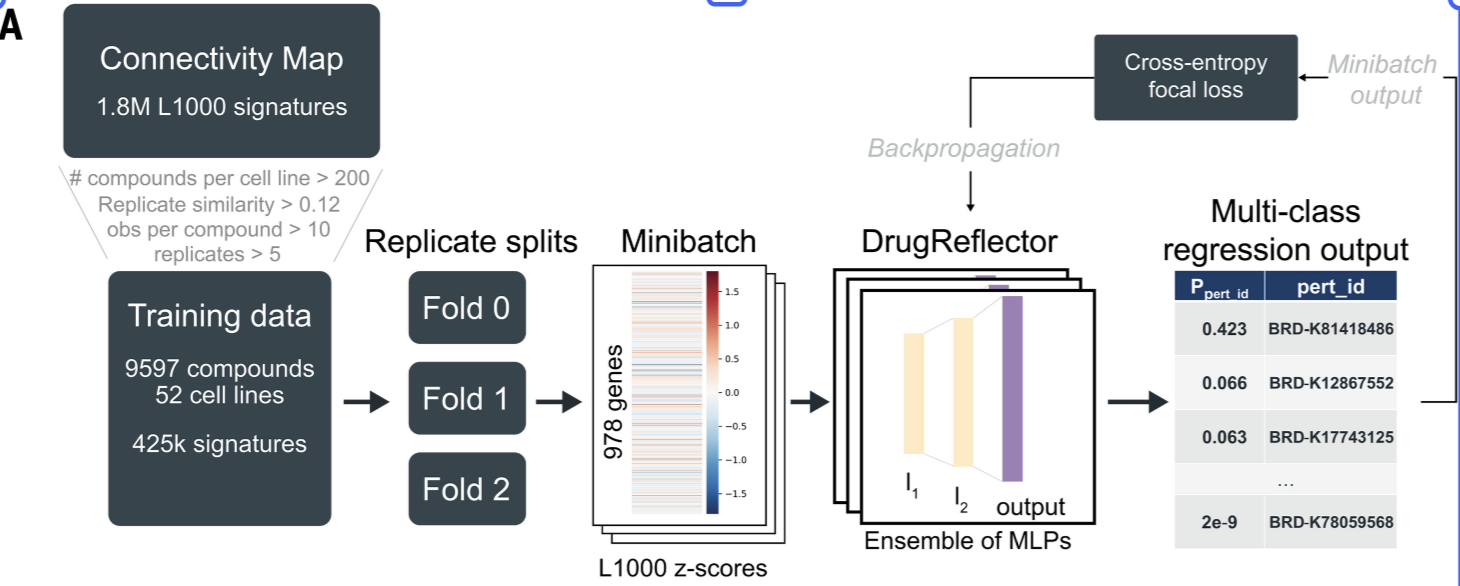
A Science paper Active learning framework leveraging transcriptomics identifies modulators of disease phenotypes (Oct 2025) introduced a drug/hit ranking framework called DrugReflector. DrugReflector is an ensemble of multi-layer perceptron (MLP) multi-class regression models trained on CMap L1000 gene expression profiles (z-scored), which capture transcriptomic signatures induced by chemical compounds. Given a transcriptomic signature derived from a phenotype of interest, DrugReflector prioritizes compounds whose perturbation signatures are predicted to reverse or modulate that phenotype, enabling the identification of candidate drugs that may have been previously overlooked. When incorperating downstream perturbed scRNA-seq experiments and phenotypic measurements, the process can iteratively refine the transcriptional signature of the hit. This creates a closed-loop, active learning process that improves hit recall and ranking performance over successive rounds. For computational biologists, the primary value of DrugReflector lies in the framework itself. It provides a systematic and data-driven approach to prioritize candidate hits based on desired transcriptomic changes, bridging large-scale perturbation data with phenotype-guided discovery.
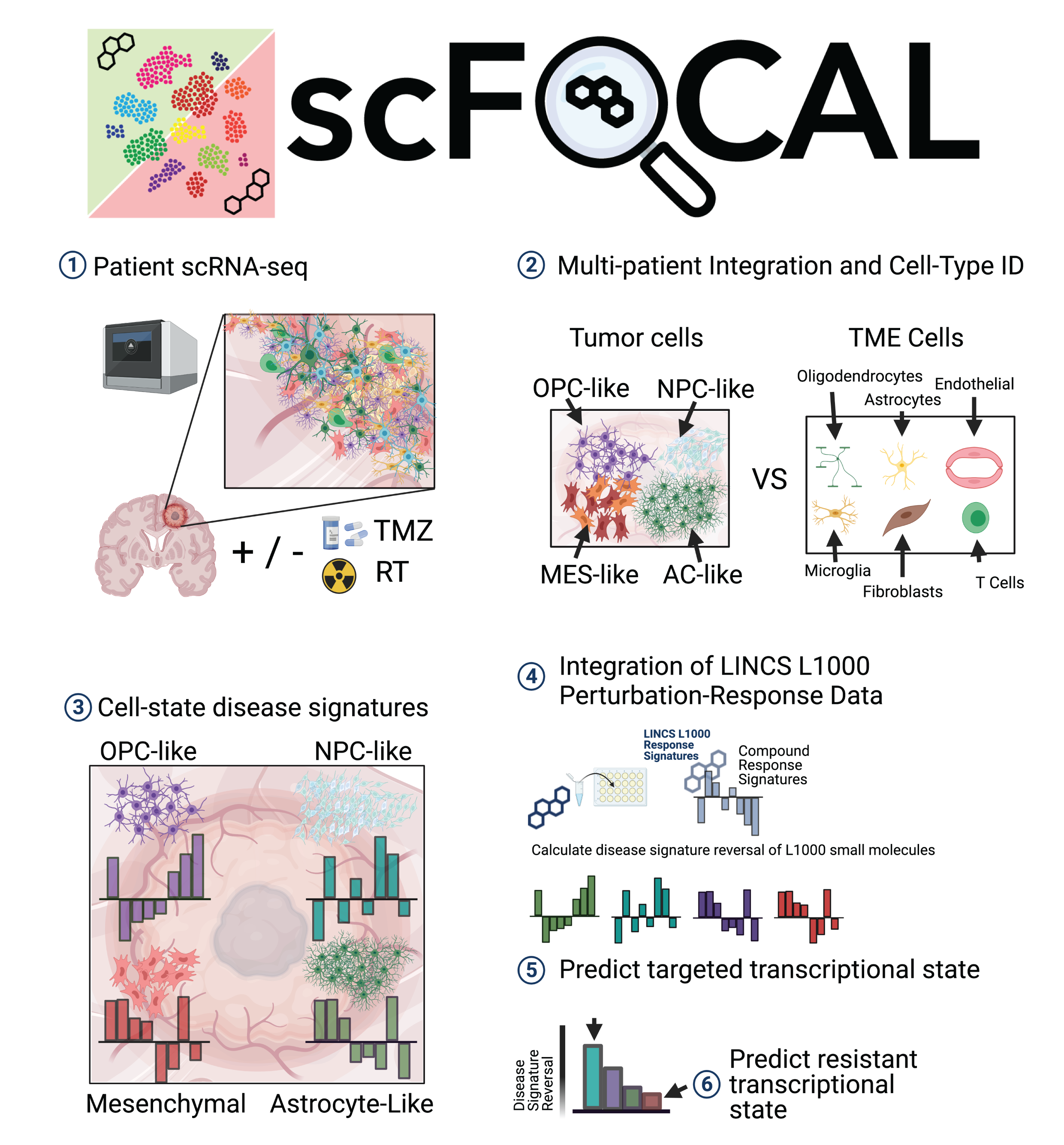
In a similar vein, a recent publication on Nature Communication introduces scFOCAL, a framework that correlates drug-response transcriptional consensus signatures (TCSs) derived from the LINCS L1000 dataset with cell-state-specific disease signature learned from multi-subject single-cell RNA sequencing data. The method predicts which cell states are targeted by a given drug and establishes drug-cell connectivity by quantifying expression discordance between perturbation and disease signature from multiple perspectives. The authors applied scFOCAL to single cell RNA-seq data from Glioblastoma (GBM) tumors and identified a combinatorial thearapautic strategy with the potential to target disease-relevant cell states in GBM.
Single-cell omics for clinic
The review by Skinnider et al. (July 2025) summarizes three major barriers limiting the clinical translation of single-cell omics: experimental, computational, and conceptual challenges.
Experimental barriers
The primary experimental limitation is the scalability of single-cell assays. When applied to large patient cohorts, single-cell omics experiments remain costly, time-consuming, and logistically complex.
In addition, technical variability, arising from sample handling, dissociation, batch effects, and platform differences, is unavoidable and complicates the identification of robust and reproducible molecular or cellular biomarkers suitable for clinical use.
Computational barriers are divided to 3 aspects:
Violation of independence assumptions: Many commonly used differential expression workflows implicitly assume that individual cells are independent observations, even when cells originate from different patients. In reality, single-cell datasets have a hierarchical (nested) structure, with cells nested within patients. Ignoring this structure leads to underestimated variance and inflated false positives. While mixed-effects models or related approaches are more appropriate for such designs, simpler methods such as cell-level Wilcoxon tests are still widely used. Pseudobulk aggregation partially mitigates this issue by restoring patient-level replication, but the review emphasizes that more principled statistical models are still needed.
Statistical “double dipping”: The authors also draw attention to a commonly overlooked issue of double dipping in single-cell analysis. Using the same dataset to (i) define cell states, clusters, or trajectories (hypothesis generation) and then (ii) test for differential gene expression between those same states or along pseudotime (hypothesis testing) can inflate false discovery rates. This practice violates fundamental assumptions of statistical inference and remains a widespread concern in exploratory single-cell workflows.
Differential abundance analysis of cell types: Changes in cell-type composition are often analyzed as if each cell type were independent. However, cell-type proportions are compositional: an increase in one population necessarily implies a decrease in others. The review points to methods such as scCODA, which adapt ideas from microbiome compositional analysis by modeling cell-type proportions jointly using a Bayesian hierarchical Dirichlet–multinomial framework, thereby accounting for uncertainty and correlation among cell types.
Conceptual barrier:
Authors argued that not all clinical questions benefit equally from single-cell omics. Single-cell approaches are most informative when disease phenotypes are driven by multicellular programs (MCPs)—-coordinated interactions among multiple cell types. For example, in cancer, tumor cells, stromal cells, and the tumor microenvironment jointly influence prognosis and therapeutic response. Methods such as DIALOGUE are highlighted as approaches that explicitly model multicellular programs by identifying coordinated gene expression patterns across interacting cell populations.
Because clinical outcomes are typically measured at the patient level, the review emphasizes the importance of integrating MCPs into patient-level predictive models. Machine learning frameworks such as multiple instance learning (MIL) are increasingly used for this purpose, treating individual cells as instances and patients as bags. Attention-based MIL models can learn which cellular states contribute most strongly to a given phenotype. MultiMIL is one such method that falls into this emerging class of approaches.
Pertpy
The last paper I want to share this week is Pertpy, an end-to-end Python-based framework for analyzing large-scale single-cell perturbation experiments, whether genetic, pharmacological, environmental, or disease-related. It is built within the scverse ecosystem, interoperating with popular single-cell tools and allowing flexible composition of analysis modules for different experimental designs and data types
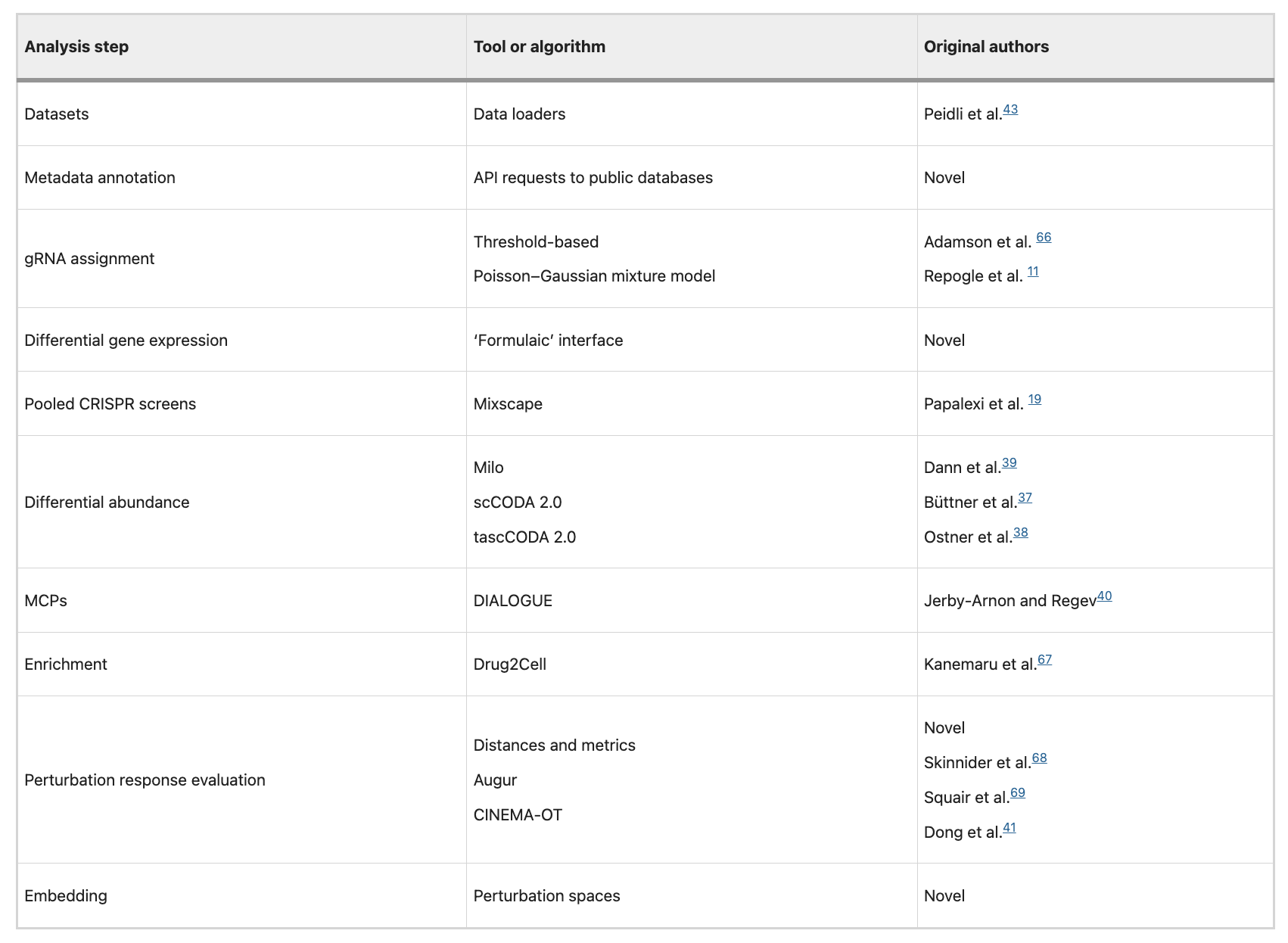
The authors demonstrate Pertpy’s versatility across several use cases:
Learning perturbation space embeddings to capture the biological similarity of perturbation effects and to cluster gene programs or perturbations based on shared responses in scRNA-seq perturbation data.
Decomposing chemical perturbation responses into viability-dependent and viability-independent components in large drug-response scRNA-seq screens by integrating annotated metadata from public resources such as DepMap, CMap, and drug databases.
Identifying compositional changes and ranking perturbation effects in complex multicellular contexts by leveraging tools like scCODA and DIALOGUE
R, data science and LLM
Polyglot in R and Python:
R + Python: From polyglot to pluralism by Emily Riederer reflects on how the R and Python ecosystems influence one another, not just co-existing but actively cross-pollinating ideas in tooling, package design, and community practices
pytest-r-snapshot, a new Python pytest plugin developed by Nan Xiao, makes it easy to snapshot unit test Python code against reference outputs generated by R. This is particularly useful when porting an R implementation to Python or ensuring feature parity between the two languages.
As an R programmer, I have started building my own cheetsheets to translate common R syntax (e.g.,
{tidyverse},{tidymodels}workflows) into popular Python equivalents for data science, such as{pandas}and{numpy}for data wrangling,{plotnine},{matplotlib}, and{seaborn}for visualization, and{scikit-learn}for machine learning. I plan to make this repository public once it is complete.
A few new tips on ggplots: 1. Joshua Marie’s new blog difference of
curly-curlyandbang-bangto use variable name insideaes2. He also listed example to usestat_afterwhich i overlooked all the time. 3. Steven Ponce’s demo to visualize Witcher 3 data demonstrates a good example to build custom theme andannotateplotClaude Code has sparked several “first impressions” from bloggers I follow1,2,3. After taking the Deeplearning.ai new course by Anthropic, I agree that tools like this are reshaping the coding experience by moving beyond autocomplete toward autonomous and challenges traditional ways we learn and practice programming. It makes the idea of AI replacing developers feel much more tangible. Instead of feeling anxious, I find myself echoing Steven Turner’s advice: “Stop telling yourself you’re behind. Recognize that you’re early.” Let’s embrace the change and learn in public.
Footnotes
My First Look at Claude Code by Stephen Turner. https://blog.stephenturner.us/p/claude-code-first-look↩︎
First impressions of Claude Cowork, Anthropic’s general agent. https://simonw.substack.com/p/first-impressions-of-claude-cowork↩︎
Claude Code and What Comes Next. https://www.oneusefulthing.org/p/claude-code-and-what-comes-next↩︎