bitBites (2026-01-07): R stats, LLM prompts and update, Virtual cell
R, data science and LLM
R Package Development Advent Calendar 2025 by Athanasia Mo Mowinckel offers a comprehensive journey through R package development. Even with prior experience, I still learned new things, especially around testthat. It led me to visit Testing chapter on R packages book. While tools like the Positron Assistant can help write unit tests, understanding the structure and principles of testthat remains essential for effective, human-aware vibe coding.
gentle introduction to mixed effects models by Nicola Romanò explains when mixed-effects models are useful, especially in designs with repeated or clustered observations where the assumption of independent samples is violated. In such cases, including random effects (instead of treating group indicators solely as fixed predictors) helps correctly model the variability at each level and avoids pseudoreplication that can inflate degrees of freedom and wrongly put random effect variable effect into “residuals”. Unlike just adding group factors as fixed effects, random effects treat group-level variation as drawn from a common distribution, allowing better generalization beyond the sampled levels and more realistic handling of hierarchical data structures. It also shows how to use
{nlme}R package to perform mixed effect linear model.Power analysis - A flexible simulation approach using R by Nicola Romanò introduces how to use simulation-based methods in R for power analysis, which goes beyond what analytical tools like the
{pwr}package can handle (ie, complex designs and mixed models where no simple formula exists). In her example, she compares two experimental designs with the same number of observations: a nested repeated-measures design analysed with a linear mixed-effects model (with individual as random effect), and an independent design analysed with a standard linear model. The simulation demonstrates that the repeated-measures design achieves higher statistical power for detecting atreatment:timeeffect with the same sample size, making it more efficient in this context.Several other R stats blogs interests me:
2025: The year in LLMs by Simon Willison provides a comprehensive overview of major developments in the LLM landscape throughout 2025. Many of the themes he highlights, including reasoning, agents, Chinese models, Google catching up, and vibe coding, strongly align with my own observations over the past year. I’ll admit that the rapid progress of LLMs caused me a fair amount of anxiety, as I worried whether computational biology might be among the next fields disrupted by AI. The creation of this bitBite series is a direct response to that anxiety. Continuing to learn and adapt feels like the only viable path forward.
Anti-hallucination prompt is fascinating. It highlights how prompt design can significantly reduce hallucinations in LLM outputs. One particularly effective strategy is the Chain-of-Verification (CoVe) method, which structures prompts so the model not only produces an initial answer but also generates and answers verification questions to check its work. An example of a grounding prompt you might use is something like: “According to [trusted source], [question]. Think through this step-by-step. If you’re uncertain about any part, explicitly state “I don’t know” rather than guessing.”
Biology and Bioinformatics
Virtual Cell Challenge 2025 by Arc Institute wrapped up in December. The challenge was to predict transcriptomic responses to CRISPRi perturbations with 300 gene KD perturb-seq data in iPSC as training set, while participates can bring their own training data and vector space to enhance the model. While there were some negative reactions on social media, mainly arguing that the concept of a “virtual cell” is too broad to be represented by a purely genetic perturbation task, these critiques are not unfounded. Drug and environmental perturbations often induce more graded and noisy responses and may better reflect a fuller notion of a virtual cell. In addition, transcriptomics alone is not a definitive representation of cellular state; proteomic and metabolomic layers are often more proximal to phenotype. That said, this deliberately simplified challenge still revealed several important lessons for building virtual cell models. First, hybrid approaches that combine deep learning with more conventional statistics consistently outperformed purely neural network–based models, and loss-function design proved to be as important as model architecture. Second, multi-modal features, especially protein-level embeddings, added measurable value. Finally, the results underscored that no single metric fully captures model quality: optimizing for one performance metric often came at the expense of others, highlighting the critical role of thoughtful metric design.
The Virtual Cell Will Be More Like Gwas Than Alphafold by Andrew Carroll echoes a similar theme. Carroll argues that building a “virtual cell” is likely to resemble GWAS. In both cases, the field must contend with noisy phenotype measurements (e.g., scRNA-seq with strong batch effects versus traits measured across heterogeneous cohorts), outcomes (transcriptomic states versus complex traits) shaped by a combination of genetic and environmental factors, and reliance on a small number of foundational datasets (large scRNA-seq atlases versus resources like UK Biobank and All-of-Us). The article outlines several key research directions needed to move toward a practical virtual cell: (1) developing methods to separate biological signal from confounding technical noise, (2) designing evaluation metrics tightly linked to clearly defined task objectives, and (3) grounding models in biological insight, with discoveries supported by orthogonal experimental or computational validation.
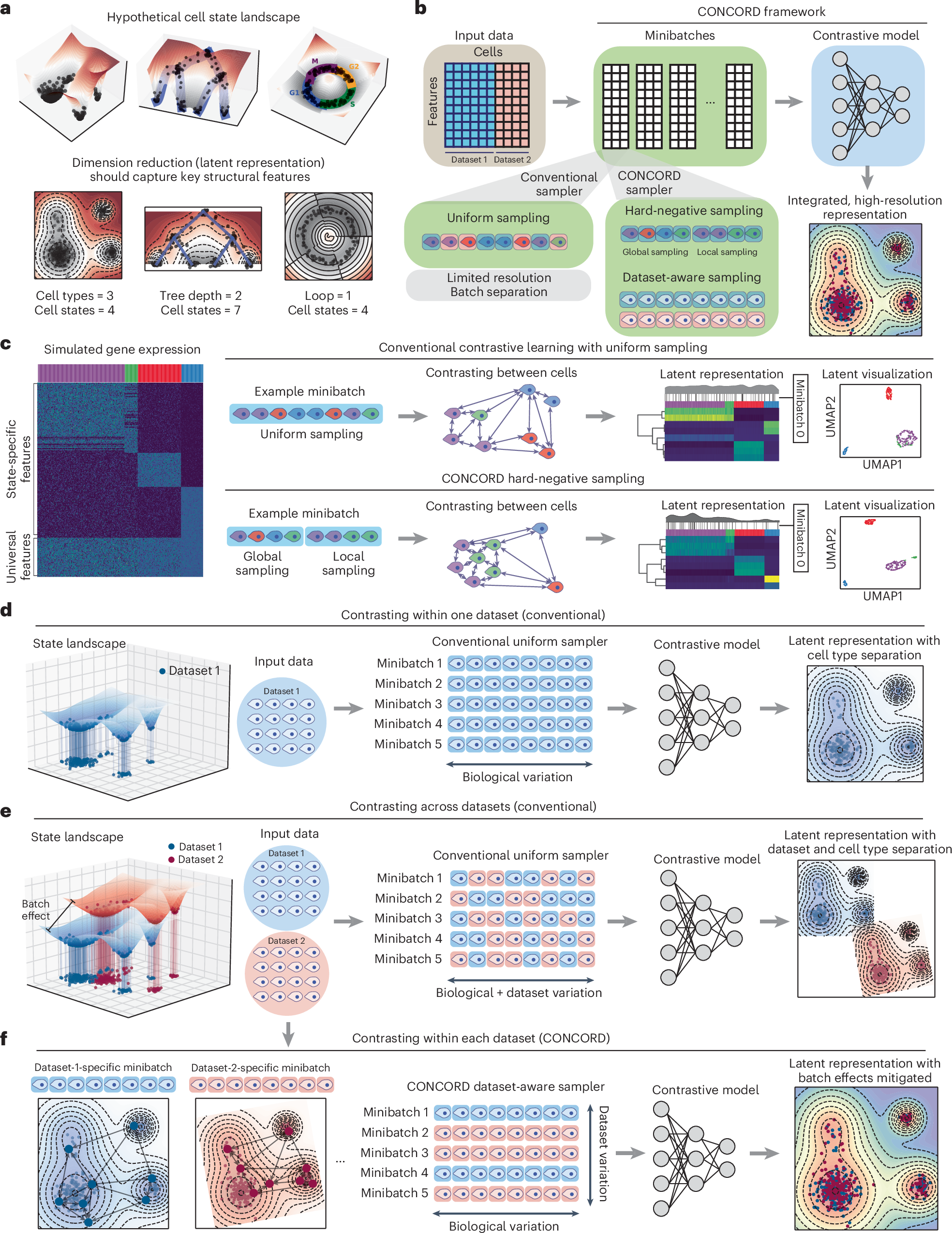
CONCORD published this month in Nature Biotechnology, describes a new single-cell integration and batch correction method that uses contrastive learning together with a probabilistic sampling strategy to learn biologically meaningful cell representations. The approach uses two complementary samplers: a data-aware sampler, which constructs minibatches enriched for cells from the same dataset to prevent technical bias between datasets, and a hard-negative sampler, which building both local (finer cell states) and global (major cell types) minibatches to describe the biological signal at different grade. This design enables two use cases relevant to my interests:
integrate data across batches, technologies, and even species, combining modalities such as spatial transcriptomics (e.g., Xenium), 3′- and 5′-based scRNA-seq, and apply to other modality (e.g., scATAC), enabling scalable construction of high-resolution cell atlases.
produces latent cell representations that capture interpretable biological structure – each latent dimension tends to reflect coherent gene-coexpression programs, and can be interpreted using gradient-based attribution techniques to link latent features back to gene and pathways that are interpretable.
github: https://github.com/Gartner-Lab/Concord
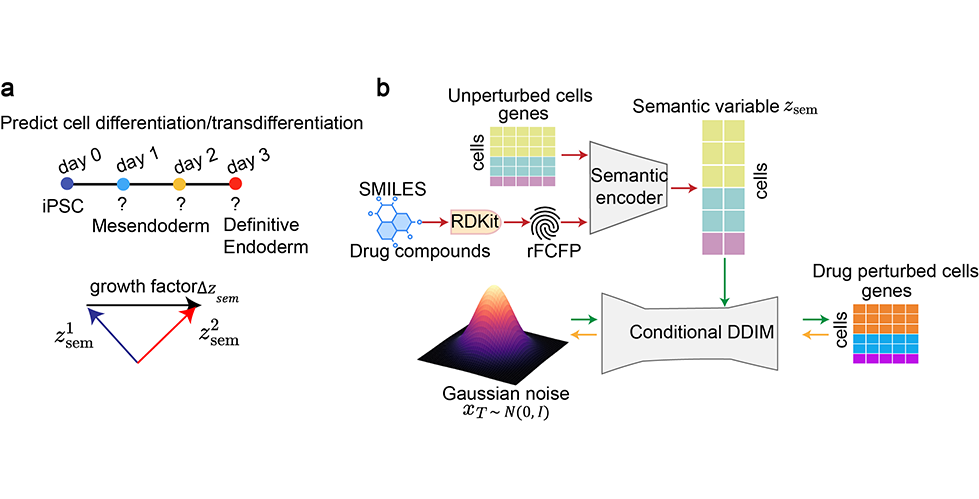
Squidiff published in Nature Methods late last year, introduces an in silico perturbation framework based on diffusion models to predict transcriptomic outcomes driven by developmental trajectories (time-course) as well as drug and genetic perturbations. Compared with previously benchmarked approaches such as GEARS and scGen, SquiDiff is better suited to modeling fine-grained, continuous, and gradient-like transcriptomic changes, which are common in developmental processes and pharmacological perturbation. This advantage stems from the diffusion framework’s ability to model smooth transitions between cellular states rather than discrete shifts. The use cases that particularly interest me include:
Predicting double-gene perturbation effects using models trained only on single-gene perturbation scRNA-seq data.
Cross-cell-type drug response prediction, where a model trained on drug-versus-control scRNA-seq in cell type A is used to predict the drug response in cell type B given only control scRNA-seq for cell type B.
interpolating intermediate cellular states in time-course experiments, by training on scRNA-seq data from the start and end time points. This is a capability that is particularly distinctive to the SquiDiff framework.
github: https://github.com/siyuh/Squidiff and https://github.com/siyuh/Squidiff_reproducibility